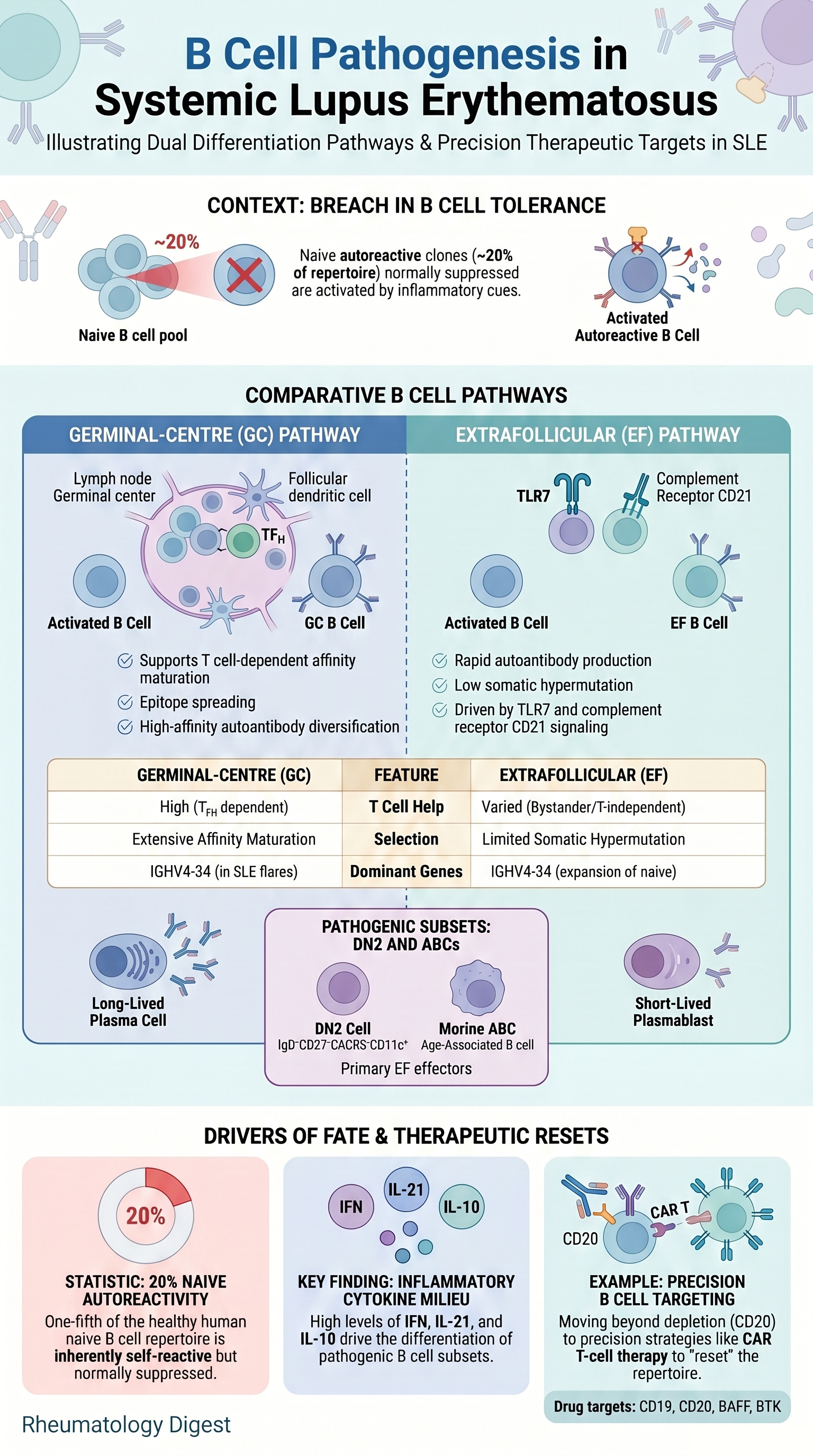
TL;DR: In SLE, autoreactive B cells are recruited into either the germinal-centre or extrafollicular pathway by their inflammatory niche — and which route they take determines autoantibody character, flare kinetics, and which therapeutic targets matter.
This review reframes how we think about autoantibody production in SLE — moving away from the B cell as an isolated autoantibody factory and towards the B cell as a cell whose fate is dictated by the inflammatory niche it inhabits.
The Clinical Problem
Autoantibody flares are central drivers of organ pathology in SLE, and they reflect a breach of B cell tolerance. The clinically inconvenient truth is that autoreactivity is normal: roughly 20% of the mature human naive B cell repertoire shows detectable self-reactivity in vitro, and autoreactive CD4+ T cells similarly circulate in healthy people. Tolerance is therefore not the absence of autoreactive clones but the active suppression of them through layered checkpoints.
In an autoimmune-prone environment, those checkpoints are lowered, and pre-existing autoreactive clones are recruited into effector pathways. Critically, once activated, an autoreactive B cell can take one of two routes — the germinal-centre (GC) pathway or the extrafollicular (EF) pathway — and these two routes produce autoantibodies with very different kinetics, affinity profiles and therapeutic vulnerabilities. The therapeutic relevance is direct: broad B cell depletion works but is non-selective. The unmet need is precision targeting of the pathogenic subset without erasing protective immunity.
What the Paper Explains
1. Intrinsic autoreactivity is the raw material
- Autoreactivity exists on a spectrum, not as a fixed threshold; some clones below the detection limit of binding assays remain functionally relevant in vivo.
- Certain autoreactive clones are actively positively selected into the mature repertoire through autoantigen engagement.
- Germline genetics matter: the heavy-chain gene segment IGHV4-34 (detected by the 9G4 anti-idiotype antibody) is disproportionately expanded across naive, memory and antibody-secreting cell (ASC) compartments during SLE flares. IGHV1-69 dominates HCV-associated cryoglobulinaemic vasculitis. The murine orthologue of IGHV4-34 (IGHV3-6) is heavily used in autoimmune chimeric mice — i.e. the same clone is “selected for” across species.
Bottom line: inherent autoreactivity does not cause disease, but it supplies the substrate that environmental cues then exploit.
2. The two pathways — a contrast worth memorising
GC pathway:
- Classical site of selection, class-switch recombination and affinity maturation.
- Not-so-obvious update: recent work shows GCs export a continuum of low-to-high affinity plasma cells — high affinity is not a prerequisite for plasma-cell differentiation. The GC is an “open structure”.
- In SLE, 9G4-positive autoreactive B cells — normally excluded from GCs in healthy tonsils — are found inside SLE germinal centres. This defective exclusion is a hallmark of the disease.
- Spontaneous autoreactive GC formation is a signature of peripheral tolerance breakdown.
EF pathway:
- Rapid: plasmablasts expand exponentially in extrafollicular foci by ~4–5 days post-immunisation; circulating ASCs appear in human blood ~4–7 days after vaccination/infection.
- Autoantibodies are lower affinity and polyreactive.
- BCR sequencing of circulating ASCs in SLE flares shows markedly reduced somatic hypermutation and marked polyclonality — consistent with antibody production largely outside GCs.
- The key human EF cell is the DN2 cell (IgD−CD27−CXCR5−CD11c+), one of the DN1–DN4 double-negative subsets; it is robustly expanded in SLE, readily becomes an ASC, and characteristically shows low/absent CD21 (CR2).
- Equivalent populations: ABCs (age-associated B cells, in aged/autoimmune mice) and atBCs (atypical B cells, in Plasmodium/Salmonella infection).
3. What decides GC versus EF fate
- Antigen availability: In infection, antigen is cleared (degraded by metalloproteases outside follicles within ~3 days; protected inside follicles by follicular dendritic cells). In autoimmunity, self-antigen is continuously abundant. High antigen density biases towards EF and can override affinity thresholds; low density favours GC. Defective clearance (DNase failure, complement C4 deficiency) tips the balance — and notably C4A versus C4B isotypes confer distinct self-reactivity patterns.
- Cytokines: IL-12 drives EF and suppresses GC (Salmonella model); IFNγ from T cells is a central driver of human DN2 development (via T-bet); IL-2 biases towards plasma cells via an mTOR–IRF4 axis; IL-21 and IL-10 modulate T cell-dependent EF help. Type I/II/III interferon signatures are a hallmark of SLE.
- Transcription/epigenetics: ZEB2 is required for ABC development and tunes the MEF2B/MEF2C balance (MEF2B is needed for GC entry). Naive B cells in SLE carry an epigenetic profile pre-disposed to EF differentiation.
- Metabolism: A genuinely elegant point — extrafollicular plasmablasts are a “nutrient sink” (l-glutamine depletion impairs GCs in malaria; supplementation rescues them). LDHA deletion in resting naive B cells abolishes GC responses while sparing EF; deletion in activated B cells does not. In COVID-19, mitochondrial dysfunction acts as a brake on EF expansion — and reduced mitochondrial dysfunction in severe disease permits pathogenic EF expansion. In SLE, the m6A demethylase FTO links TLR7 signalling to mitochondrial oxidative phosphorylation, skewing B cells towards DN2 fate.
4. The autoreactive germinal centre in SLE
- GCs generate autoreactivity two ways: by admitting pre-existing self-reactive clones, and by allowing initially innocent clones to acquire self-reactivity through somatic hypermutation without effective purging.
- Autoreactive GCs are open structures that recruit wild-type B cells, driving epitope spreading (broadening of antigenic targets).
- Counter-intuitive but important: GCs amplify and diversify autoreactivity but are not required for self-reactive ASC production — B cell-specific BCL6 deletion does not reduce self-reactive ASCs and instead enhances the EF pathway.
- TFH cells adopt a permissive role; continuous T cell help is needed to sustain autoreactive GCs. Autoreactive GCs are proposed to originate from an initiator clone in the splenic extrafollicular bridging channel. Clonal redemption (mutating away from self-reactivity) is one proposed reason the system tolerates self-reactive clones entering GCs.
5. T cell help in the extrafollicular pathway
EF responses are T cell-dependent, but the dependence is flexible and TLR-calibrated. EF help is ICOS-dependent yet SAP-independent — meaning the stable, long-lived T–B contacts essential for GCs are less essential extrafollicularly.
In the AM14 model, TLR7/TLR9 signalling can drive EF activation and even somatic hypermutation independently of T cells, with CD4+ T cells amplifying rather than dictating the response.
Distinct EF helper T cell subsets are emerging:
- T_PH cells (T peripheral helper; PD1hiCXCR5−CD4+) — IL-21, CXCL13, SAP; first described in RA synovium, correlate with CD11c+ B cells in SLE.
- T_H10 cells (CXCR5−CXCR3+PD1hi) — IL-10- and succinate-driven, IL-21-independent; enriched in paediatric lupus nephritis tubulointerstitium; amplified by oxidised mtDNA-activated plasmacytoid dendritic cells.
- T_HA cells (age-associated T helper) — ZEB2/T-bet-driven, age-dependent, with cytotoxic features (granzymes, perforin).
A practical species caveat: trimerised CD40L activates resting naive human B cells but not DN2 cells — DN2 cells are instead highly TLR7-responsive, indicating reduced reliance on cognate T cell help.
6. Memory and therapy
- Conventional switched memory B cells (IgD−CD27+) appear largely unaffected in SLE.
- Important reframing: DN2 cells are likely not a stable memory pool — they arise from naive precursors and behave as immediate ASC progenitors. The “atypical memory B cell” label may be misleading.
- B cell-targeted therapy: anti-CD20/CD19/BAFF and BTK inhibitors show variable efficacy. CD19- and BCMA-directed CAR T cells show promise, including an immunological “reset” with non-autoreactive reconstitution; self-reactive antibodies and CD11c+CD21− B cells were minimal at 6–12 months, though a modest memory B cell rebound was seen at 1 year. In TLR7 gain-of-function mice, post-CAR-T relapse after CD19 targeting was driven mainly by newly generated B cells, whereas BCMA-targeted relapse had mixed origin.
- Because naive B cells in SLE carry epigenetic imprinting predisposing to activation, peripheral depletion may not erase the underlying defect — supporting non-depleting strategies (targeting the BCR complex, CD19, IFN receptors, CD21, or IGHV4-34-specific CAR T cells).
Key Takeaways
- Autoreactivity is normal; disease is a fate decision. The pathogenic event in SLE is not the existence of autoreactive clones but the environment that recruits them into effector pathways.
- GC and EF pathways are complementary, not mutually exclusive. The GC diversifies autoreactivity and drives epitope spreading; the EF pathway provides rapid, low-affinity, polyclonal autoantibody output. EF responses often precede GC responses rather than excluding them.
- The germinal centre is “open” and permissive in SLE. Loss of 9G4/autoreactive B cell exclusion is a disease hallmark — and GCs are dispensable for self-reactive ASC generation (BCL6 deletion shifts output to EF).
- Fate is set by the niche. Antigen abundance, cytokines (especially IFNγ and IL-12), epigenetic imprinting and — strikingly — metabolic resources and mitochondrial state all bias the GC-versus-EF decision.
- EF T cell help is heterogeneous and TLR-calibrated. T_PH, T_H10 and T_HA cells are distinct emerging helper subsets; TLR7 signalling can substitute for, or reduce reliance on, cognate T cell help.
- DN2 cells are progenitors, not memory. This distinction matters for interpreting biomarkers and for predicting which cells survive depletion therapy.
- Future therapy = precision, not erasure. The conceptual goal is to redirect or reversibly modulate the route of autoantibody production — sparing protective immunity — rather than depleting the entire repertoire. Long-term monitoring of reconstituting B cell and T cell clonality (and interferon activity) after CAR T is essential.
Final Take-Home for Practice
For the practising rheumatologist, the central message is conceptual: think of B cells in the context of their inflammatory milieu, not as isolated antibody producers. The variability we see clinically — flare tempo, autoantibody breadth, paediatric versus adult organ patterns, and response to depletion — plausibly maps onto whether a patient’s autoantibodies are being generated through GC or EF routes. As IFN-targeted agents, CAR T platforms and (eventually) pathway-selective biologics mature, distinguishing these two pathways may move from being an academic distinction to a therapeutically actionable one.