TL;DR: Morphea is an organ-sparing autoimmune skin disease — fundamentally different from systemic sclerosis — where depth and site dictate everything: deep, linear, facial or joint-overlying disease needs immediate long-course systemic therapy (methotrexate + a steroid bridge, given >24 months), never watchful waiting, because treatment delay is the strongest driver of irreversible damage and relapse.
The Clinical Problem
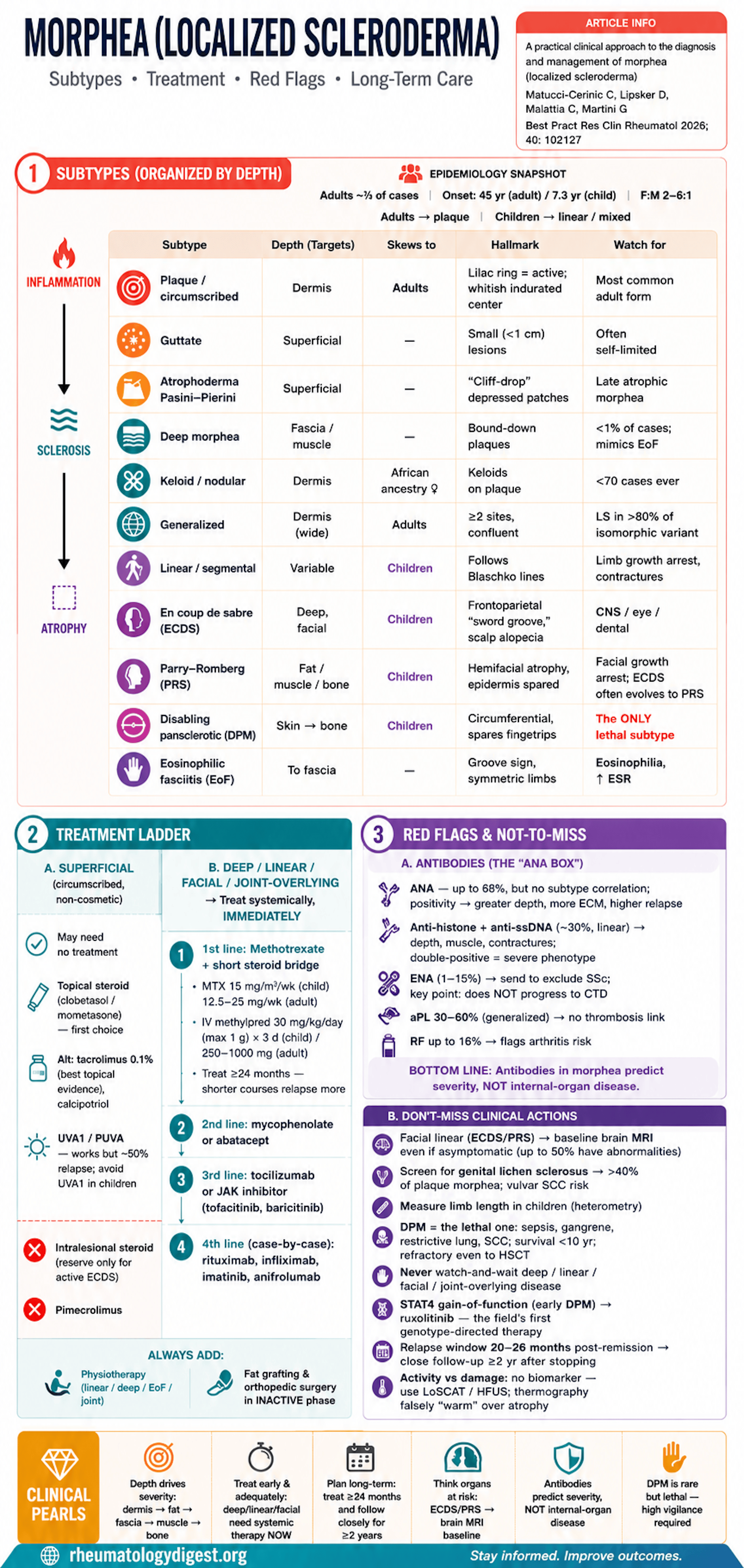
Morphea (localized scleroderma, LS) is a heterogeneous group of inflammatory/fibrosing skin disorders that progress through inflammation → sclerosis → atrophy, with severity governed by the depth of tissue involvement (dermis → fat → fascia → muscle → bone).
The authors are deliberate about terminology: they prefer “morphea” over “localized scleroderma” precisely because the latter is confused with systemic sclerosis (SSc) — and the two are fundamentally different diseases.
The core clinical challenges:
- Diagnostic delay is the rule, not the exception — onset is insidious, the differential is broad, and there are no validated biomarkers of disease activity.
- Distinguishing active disease from irreversible damage remains genuinely difficult at the bedside and drives both under- and over-treatment.
- In children especially, delayed or inadequate therapy of linear/deep forms causes irreversible cosmetic, orthopedic, and functional damage (growth arrest, contractures, facial asymmetry).
Two crucial framing points that separate morphea from SSc:
- Morphea predominates in childhood and almost never involves internal organs — even when autoantibodies are present.
- It is nonetheless a true autoimmune disease, with increased overlap with other autoimmune conditions.
Classification — and Why It Keeps Changing
Four systems coexist (Peterson 1995, Padua/juvenile 2006, EDF 2017, German S2K 2024). The repeated revisions reflect how poorly a heterogeneous, overlapping disease fits rigid phenotypic boxes.
- Non-obvious but high-yield point: when the three older systems were tested head-to-head, the Padua criteria classified 95% of patients vs only 56% (Peterson) and 52% (EDF).
- The main point of disagreement is where eosinophilic fasciitis (EoF) belongs — separate entity vs a “special type” of linear morphea (S2K).
Broad subtype families: circumscribed/plaque (limited), generalized, linear/segmental, deep, pansclerotic, mixed, plus EoF.
Epidemiology
- Adult incidence 4–27 / million/yr; pediatric 2–3.4 / million/yr; ~two-thirds of cases are adults.
- Mean onset 45 yrs (adults) vs 7.3 yrs (children); neonatal onset described.
- Female predominance 2–6:1.
- Subtype by age: adults → plaque (then generalized); children → linear and mixed.
Clinical Spectrum — the Subtypes Worth Teaching
- Plaque/circumscribed: oval lesions, erythema → indurated whitish center; the active “lilac ring” halo is the key activity sign; late lesions soften, atrophy, dyspigment.
- Guttate: small (<1 cm) superficial lesions; often self-limited.
- Atrophoderma of Pasini–Pierini: “cliff-drop”/canoe-like depressed hyperpigmented patches; histologically a late atrophic morphea.
- Deep morphea: rarest limited variant (<1% of cases); reaches fascia/muscle, can cause functional limitation; hard to distinguish from EoF.
- Keloid (nodular): very rare (<70 cases), mainly middle-aged women of African ancestry.
- Generalized: confluent plaques over ≥2 sites (definitions vary). Non-obvious: genital lichen sclerosus is present in >80% of the isomorphic variant.
- Disabling pansclerotic morphea (DPM): the dangerous one — circumferential skin-to-bone involvement, contractures, ulceration, typically spares fingertips, often childhood onset. The only morphea subtype with significant mortality (sepsis, gangrene, restrictive lung disease, SCC; post-diagnosis survival <10 years), and it is refractory even to autologous stem cell transplant.
- Linear/segmental: follows Blaschko lines; in children damages limb growth/trophism → flexion contractures.
- En coup de sabre (ECDS): frontoparietal sclerotic groove (“sword wound”); scalp alopecia; can extend to eye/oral region. May initially mimic port-wine stain or lichen striatus.
- Parry–Romberg syndrome (PRS, progressive hemifacial atrophy): deep atrophy of fat/muscle/bone with epidermis usually spared; childhood onset → facial growth arrest, dental malocclusion. A substantial proportion of ECDS evolves into PRS.
- EoF (Shulman): skin-to-fascia involvement, groove sign, symmetric limbs; blood eosinophilia, raised ESR, hypergammaglobulinemia early. Children → better prognosis, less arthritis, and none of the adult hematologic associations (aplastic anemia, thrombocytopenic purpura, malignancy).
The single most important new mechanistic insight: In four children with very-early-onset DPM (mucosal ulcers + sclerosis), heterozygous gain-of-function STAT4 mutations were found; their fibroblasts showed impaired wound healing and very high IL-6, and they responded to the JAK inhibitor ruxolitinib. This is described as the only known causative mutation in the entire sclerodermatous-disease spectrum — a clean genotype-to-targeted-therapy story.
Laboratory Findings — Generally Unhelpful, with Caveats
- Routine bloods (CBC, renal/liver, CK, urinalysis) are usually normal. EoF is the exception (eosinophilia, high ESR).
- ANA positive in up to 68% — but no correlation with subtype; a meta-analysis links ANA to greater depth, more extracutaneous disease, and higher relapse.
- Anti-histone + anti-ssDNA (~30%, esp. linear) correlate with depth/muscle/contractures; double positivity = more severe phenotype.
- ENA antibodies appear in 1–15% but do not predict subtype and — importantly — do NOT progress to the corresponding connective-tissue disease.
- Antiphospholipid antibodies in 30–60% (esp. generalized) but no thrombotic correlation. RF up to 16% → flags arthritis risk (esp. generalized).
Extracutaneous Manifestations — a Pediatric, Linear-Disease Problem
- Rare in adults; ~1 in 5 children affected. In 750 jLS patients: articular 47%, neurologic 17%, vascular 9%, ocular 8%, GI 6%, respiratory, cardiac, renal each ≤2.6%.
- Linear (facial) morphea drives most ECM.
- CNS (seizures, headache/migraine, behavioral/intellectual changes, neuropathy) clusters with ECDS/PRS — and up to 50% of asymptomatic ECDS/PRS patients have brain MRI abnormalities (T2 white-matter lesions, leptomeningeal enhancement, calcifications, rarely vasculitis). Hence baseline brain MRI is recommended in all facial linear disease, symptomatic or not.
- Ocular findings (adnexal changes, uveitis, episcleritis) and odontostomatologic complications need dedicated specialist follow-up.
Diagnostic Approach (Clinical First)
- Diagnosis is clinical; biopsy only when uncertain (depth to fat for linear/deep, to fascia for EoF).
- Mandatory checks people forget: genital exam/inquiry for lichen sclerosus (associated >40% of plaque morphea, with vulvar SCC risk); TMJ and articular exam; limb length measurement in children to catch heterometry; ophthalmology at baseline + follow-up; brain MRI + maxillofacial review for facial disease.
- ENA should be sent — not to subtype morphea, but to exclude SSc. Authors also suggest screening coeliac and thyroid disease; Borrelia only in endemic/suspicious cases.
Activity vs Damage Assessment Tools
- LoSCAT (validated): combines PGA + mLoSSI (activity: erythema, thickness, new/extending lesions) + LoSDI (damage: atrophy, fat loss, dyspigmentation). Newer LoTSS adds extracutaneous burden.
- Infrared thermography: sens/spec ~80.7% / 86.3% — but false “warm” readings in atrophic lesions, so avoid with marked atrophy.
- High-frequency US (≥15 MHz): recommended for activity/extent/response; newer SMI (microvascular Doppler) and shear-wave elastography (operator-independent) are emerging.
- MRI: best for deep/linear/EoF (joint, muscle, bone, length discrepancy, surgical planning); poor for superficial layers, costly, may need sedation.
Management — the Therapeutic Spine
The unifying rule: depth and site dictate aggressiveness.
- Superficial circumscribed lesions in non-cosmetic areas: may need no treatment, or topicals.
- Deep / linear / cosmetic-area / joint-overlying lesions: treat immediately and systemically — do not “watch and wait.”
Topical (superficial disease):
- High-potency steroids (clobetasol) or medium-potency (mometasone); intralesional steroids not recommended (reserve only for active ECDS).
- Alternatives: topical tacrolimus 0.1% (best evidence among topicals), calcipotriol (± steroid/phototherapy); pimecrolimus not recommended.
- UVA1 / PUVA phototherapy effective but ~50% relapse; avoid UVA1 in children (photoaging/carcinogenesis risk).
Systemic (deep/linear/generalized/EoF):
- First line: methotrexate + a short systemic-steroid bridge. MTX backed by RCT and cohort data; dose 15 mg/m²/wk (children) or 12.5–25 mg/wk (adults). Key durability point: relapse is higher if treated <24 months, so treat long.
- Steroids: IV methylprednisolone 30 mg/kg/day (max 1 g) ×3 days (children) / 250–1000 mg (adults), or oral prednisone with taper — as a bridge, not monotherapy.
- Second line: mycophenolate (add-on or switch) or abatacept (notably effective even in pansclerotic/generalized and on arthritis).
- Third line: tocilizumab (>40 reported LS cases, incl. pansclerotic) or JAK inhibitors (tofacitinib, baricitinib; topical ruxolitinib/delgocitinib) — promising, not yet in guidelines.
- Fourth line / case-by-case: rituximab, infliximab, imatinib, anifrolumab.
- Adjuncts: physiotherapy is mandatory in linear/deep/generalized/EoF/joint-overlying disease; autologous fat transfer (aesthetic + local anti-inflammatory/anti-fibrotic) and orthopedic surgery/epiphysiodesis in the inactive phase or under immunosuppression.
Outcome & Prognosis
- Except pansclerotic, morphea is rarely life-threatening — but the burden is functional, cosmetic, and psychological, often relapsing.
- Only ~25% remit (children 27% > adults 17%).
- Relapse risk factors: onset age, linear-extremity and generalized subtypes, delayed treatment, ANA positivity. Relapse typically occurs 20–26 months after remission → close follow-up for ≥2 years after stopping therapy.
- Treatment delay → more relapse and damage — the recurring theme.
Key Takeaways
- Morphea ≠ systemic sclerosis. It is autoimmune but essentially organ-sparing; autoantibodies (even ENAs) carry no progression risk to CTD. The job is to protect skin, growth, and function — not internal organs.
- Depth determines everything — classification, imaging choice, and treatment aggressiveness. Deep, linear, facial, or joint-overlying disease = immediate systemic therapy, never watchful waiting.
- Don’t miss the high-stakes subtypes: DPM (the only one with real mortality, fingertip-sparing, often refractory) and facial linear disease (ECDS/PRS) (CNS, ocular, dental, growth complications — baseline brain MRI even if asymptomatic).
- Activity vs damage is the central unsolved problem — no biomarker exists; rely on LoSCAT, HFUS, and thermography (mind its atrophy pitfall).
- Methotrexate + steroid bridge is first-line systemic therapy, given long (>24 months) to prevent relapse; escalate to MMF/abatacept, then tocilizumab/JAK-i.
- Targeted therapy is arriving: the STAT4 gain-of-function → high IL-6 → ruxolitinib story in early DPM is the field’s first genotype-directed treatment, and the broader IL-6/JAK rationale underpins tocilizumab and JAK-i use.
- Screen for the silent companions: genital lichen sclerosus (>40% of plaque morphea; vulvar SCC risk) and, in children, limb-length discrepancy.
- Early diagnosis and early treatment are the strongest modifiable determinants of outcome — delay drives irreversible damage and relapse.