TL;DR: The near-term needle-movers in PsA converge on biologic-like, multidomain, increasingly oral therapy — IL-17 innovation (dual A/F blockade plus engineered formats), selective oral TYK2 inhibition, and oral IL-23R antagonism — while the more ambitious frontier is intercepting disease at the psoriasis-to-PsA transition, a plausible but still unproven strategy.
The Clinical Problem
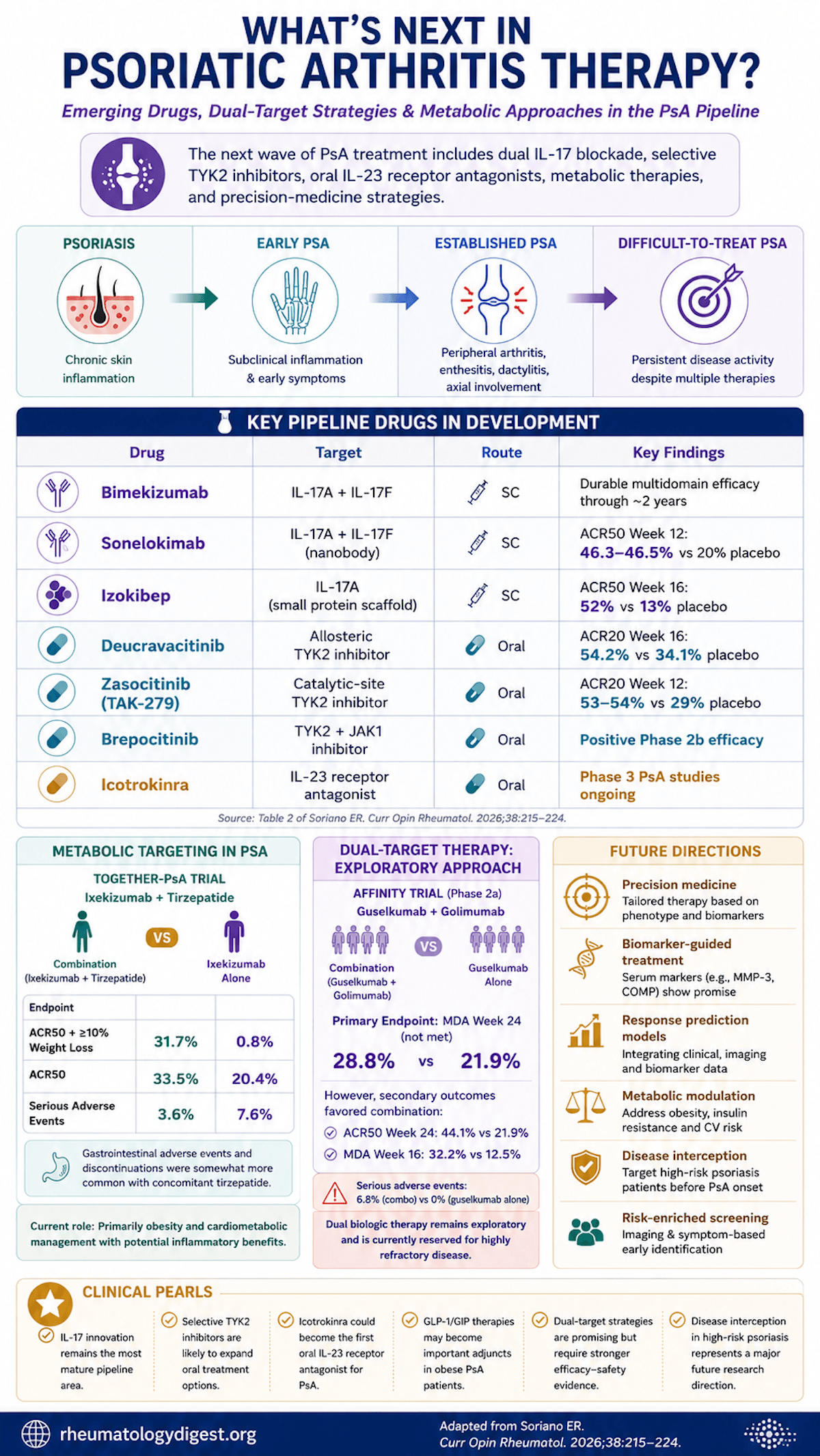
PsA is a multidomain disease — peripheral arthritis, enthesitis, dactylitis, axial involvement, skin and nails — and that breadth is exactly why it lags behind psoriasis therapeutically. In PsO, near-complete skin clearance is now a realistic target; in PsA, a meaningful share of patients never reach minimal disease activity (MDA) or low disease activity, and real-world drug survival is eroded by partial response, adverse events and access barriers. A growing subset now meets formal difficult-to-treat (D2T)/treatment-refractory definitions (recent EULAR and GRAPPA consensus work).
The review frames the pipeline around two vectors, the conceptual spine of the whole piece:
- Greater precision — higher pathway selectivity (TYK2), broader neutralisation within a pathway (IL-17A and F), engineered molecular formats (nanobodies, small scaffolds), and a decisive shift toward oral targeted therapy.
- Moving earlier — intercepting at the psoriasis-to-PsA transition before disease becomes clinically classifiable.
Prevention and Interception — the “Skin-to-Joint Window”
- Only ~30–40% of psoriasis patients develop PsA, so any interception strategy must be risk-enriched to avoid overtreating the majority who never progress.
- Risk enrichers: obesity/weight gain (modifiable), nail disease, higher PsO severity, and musculoskeletal symptoms — especially arthralgia.
- The staged model (PsO at higher risk → subclinical phase → overt PsA) maps onto two complementary signals: arthralgia marks a symptomatic prodrome, while power-Doppler enthesitis on US and MRI inflammation mark a subclinical inflammatory phase.
Do psoriasis therapies prevent PsA? The honest answer is “signal, not proof,” and the methodological caveat is the real teaching point. Observational cohorts are dogged by confounding by indication and channeling bias — biologic choice tracks the skin-vs-joint phenotype at baseline. Illustratively:
- Several propensity-matched datasets show HR ~0.3 for incident PsA with biologics.
- Cho 2025 (Korea, 9499 PsO): any IL-inhibitor vs TNFi HR 0.40; IL-23i 0.22, IL-17Ai 0.47, IL-12/23i 0.46.
- Strober 2024 (USA, 7144 PsO): incidence/100 PY — 4.99 IL-23i, 7.29 IL-17Ai, 6.06 IL-12/23i, 9.39 TNFi.
- But channeling cuts both ways: Meer (USA) found biologic-treated patients had higher incident PsA (HR 4.5), and Watad found biologics protective vs MTX (0.46) yet “harmful” vs topicals (2.16) — because sicker skin gets biologics. Direction of effect can flip with the comparator.
The PAMPA trial (guselkumab in a risk-enriched PsO cohort) is the prospective study positioned to convert these signals into causal evidence.
IL-17 Pathway Innovation — Broader Blockade + New Formats
- Bimekizumab (dual IL-17A/F mAb, SC): phase 3 ACR20 met, sustained to ~2 years. Mechanistic pearl: because IL-17F also contributes to mucosal defence, dual A/F blockade brings more mucocutaneous candidiasis as an anticipated, monitorable AE.
- Sonelokimab (nanobody, IL-17A/F, albumin-binding to extend half-life and aid tissue delivery): ARGO phase 2 — ACR50 at wk12 46.3–46.5% vs 20.0% placebo (P=0.0006), improving through wk24.
- Izokibep (small-protein scaffold, IL-17A): phase 2 — ACR50 at wk16 52% vs 13% placebo; MDA 39–42% vs 5%.
Conceptual thread: nanobodies/scaffolds are smaller than conventional mAbs, hypothesised to improve tissue penetration and target engagement — but whether that translates into superior enthesitis/axial performance is unproven and needs larger trials.
Selective TYK2 Inhibition — a Selectivity Spectrum, Not a Single Drug
TYK2 sits downstream of IL-12/IL-23 and contributes to type I IFN — so selective TYK2 blockade is conceptually closer to IL-23/12 modulation than to broad JAK inhibition. The review lays out a gradient of selectivity:
- Deucravacitinib (allosteric TYK2, oral; approved in PsO): POETYK PsA-1 — ACR20 wk16 54.2% vs 34.1%; PASI75 40.9% vs 15.4%; MDA 25.6% vs 14.7%. PsA-2 sustained to wk52; enthesitis/dactylitis improved. Herpes zoster uncommon and none in PsA trials to date.
- Zasocitinib (TAK-279) (highly selective catalytic-site TYK2, oral): phase 2b — ACR20 wk12 53.3–54.2% vs 29.2% (P=0.002); ACR50 26.4–26.7% vs 9.7%; MDA 29.2% vs 12.5% (30 mg).
- Brepocitinib (dual TYK2/JAK1, oral): phase 2b primary met; broader footprint may add efficacy in some phenotypes but reintroduces JAK-class safety/monitoring — its positioning hinges on that trade-off.
The Potentially Disruptive One — Oral IL-23R Antagonism
- Icotrokinra (JNJ-77242113): an oral peptide IL-23R antagonist with strong PsO proof-of-concept, now in phase 3 PsA across biologic-naïve and biologic-experienced populations. If joint/enthesitis/dactylitis efficacy matches its skin performance, pathway-specific oral therapy with biologic-like effect could reshape early sequencing.
Adjunctive and Strategy-Level Moves
Metabolic targeting (GLP-1 / dual incretin):
- Obesity and insulin resistance are common and predict lower MDA attainment and higher discontinuation; weight loss improves activity.
- TOGETHER-PsA (phase 3b, ixekizumab + tirzepatide, open-label/blinded assessor): primary composite (ACR50 + ≥10% weight loss) 31.7% vs 0.8% with ixekizumab alone (P<0.001); ACR50 alone 33.5% vs 20.4%; SAEs fewer with the combination (3.6% vs 7.6%), GI AEs more frequent with tirzepatide. Caveat: a composite that bundles weight loss will favour the weight-loss arm on that component — the standalone ACR50 is the more honest joint signal.
- Positioning: GLP-1RAs are comorbidity-targeted (obesity/diabetes/CV risk) with possible secondary anti-inflammatory benefit — not yet disease-modifying for PsA.
Dual biologic / dual-targeted therapy (for D2T disease):
- Real-world data (Wu, JAMA Dermatol 2025): combination is uncommon and not associated with higher serious/opportunistic infection vs monotherapy — but low certainty.
- AFFINITY (phase 2a, guselkumab + golimumab, prior TNF-IR): primary endpoint MDA wk24 NOT met (28.8% vs 21.9%; OR 1.4, 90% CI 0.6–3.3). Secondaries numerically favoured combination (ACR50 wk24 44.1% vs 21.9%; MDA wk16 32.2% vs 12.5%), enthesitis resolution similar. The trial was enriched for systemic inflammation (CRP ≥0.3 mg/dL), testing the hypothesis that higher inflammatory burden amplifies the value of dual blockade. SAEs 6.8% (combo) vs 0% (mono) — a real safety flag at this sample size.
- Sequential IL-17/IL-23 inhibition (alternating upstream/downstream) is proposed but remains case-based and hypothesis-generating.
Precision medicine: predictors today are mostly clinical (skin-vs-MSK phenotype, enthesitis/axial, obesity, central sensitisation, CRP). Soluble biomarkers (MMP-3, COMP for TNFi response; Chandran) offer proof-of-concept, but few replicate consistently; prospective stratified testing and assay standardisation are the gaps.
Key Takeaways
- The near-term needle-movers are three: IL-17 innovation (dual A/F + engineered formats), selective oral TYK2 inhibition, and oral IL-23R antagonism — converging on the same goal of biologic-like, multidomain, increasingly oral therapy.
- Think of TYK2 as a selectivity dial, not one drug: allosteric (deucravacitinib) → highly selective catalytic-site (zasocitinib) → broader dual TYK2/JAK1 (brepocitinib), with efficacy-breadth traded against safety/monitoring.
- Format matters but isn’t proven: nanobodies/scaffolds promise better tissue delivery; the open question is whether that yields superior enthesitis/axial outcomes.
- Interception is plausible but unproven — the 30–40% progression rate demands risk-enrichment, and the observational “prevention” signals are heavily confounded by channeling. Watch PAMPA.
- Mechanistic toxicity links are predictable: dual IL-17A/F → more candidiasis; broader JAK footprint → more monitoring. Use them to counsel and select.
- Strategies, not just molecules: GLP-1/incretins as comorbidity adjuncts (not yet disease-modifying), dual-targeted therapy reserved for refractory multidomain disease pending safety data, and phenotype/biomarker-informed sequencing as precision medicine matures.
- Read the endpoints critically: AFFINITY missed its primary despite favourable secondaries (and an inflammation-enriched design), and TOGETHER-PsA’s headline rests partly on a weight-loss composite — both are reminders to separate marketing-friendly composites from domain-specific joint efficacy.