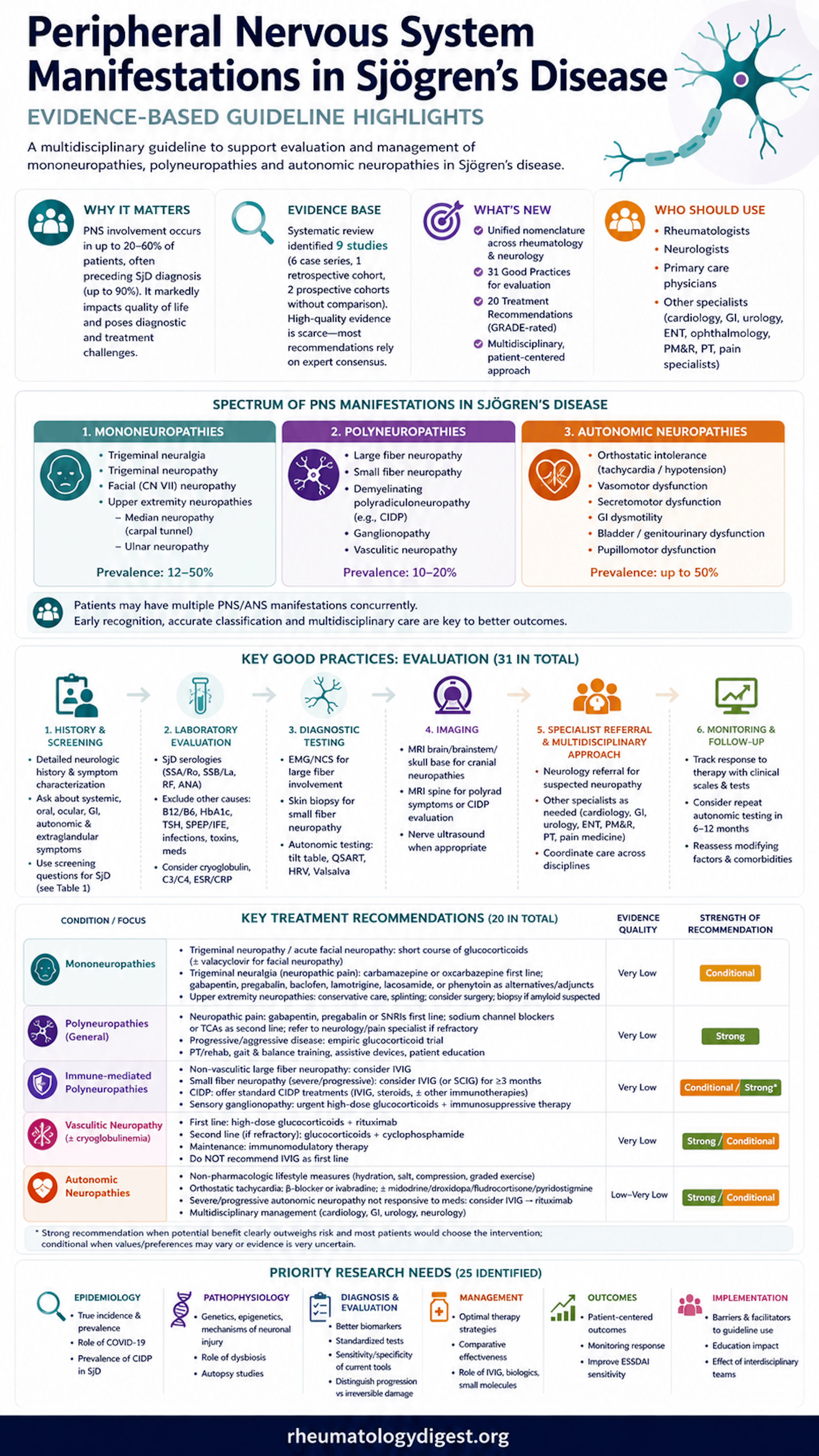
TL;DR: The first Sjögren’s Foundation guideline for peripheral nervous system disease aligns neurology–rheumatology nomenclature and issues 20 treatment recommendations (6 strong) and 31 evaluation good-practice statements — the practical spine being: suspect PNS involvement early (it often precedes diagnosis and can occur despite negative anti-SSA), characterise the subtype because treatment diverges sharply, and don’t miss vasculitic neuropathy or ganglionopathy, which need immediate aggressive immunotherapy.
The Clinical Problem
Sjögren’s disease (SjD) is a systemic autoimmune disorder that reaches well beyond sicca. Peripheral nervous system (PNS) involvement is common, under-recognised, and frequently the presenting feature — the mean age at PNS diagnosis is around 59 years, and neuropathy precedes the SjD diagnosis in as many as 90% of affected patients. Reported prevalence spans a wide range (8–60%) and is thought to be underestimated because early or asymptomatic neuropathies are missed.
A few points that are easy to overlook but clinically important:
- Roughly 30–40% of SjD patients are anti-SSA negative, so a neuropathy work-up should not be abandoned simply because SSA is absent.
- PNS manifestations frequently co-occur — a single patient may harbour more than one neuropathy type simultaneously.
- A persistent terminology gap exists between neurology and rheumatology, which historically hampered joint management. Bridging this was a core aim of the guideline.
The PNS spectrum covered here includes:
- Mononeuropathies — trigeminal neuralgia/neuropathy, facial (CN VII) neuropathy, median (carpal tunnel) and ulnar neuropathy (prevalence 12–50%).
- Polyneuropathies — large-fibre, small-fibre, demyelinating polyradiculoneuropathy (AIDP/CIDP), sensory ganglionopathy, and vasculitic neuropathy (prevalence 10–20%).
- Autonomic (ANS) neuropathies — orthostatic intolerance, POTS, orthostatic hypotension, sudomotor/GI/GU dysfunction (up to 50%).
The Aim
To produce a rigorous, multidisciplinary, evidence-based resource for the evaluation and management of PNS manifestations in SjD, structured around two overarching PICO questions: (1) which therapies most effectively improve neurologic symptoms/function in SjD-related PNS and ANS disease, and (2) which diagnostic/monitoring strategies are clinically useful for diagnosis and tracking of these manifestations.
How the Guideline Was Developed
- A Topic Review Group (TRG) was assembled: 6 neurologists, 6 rheumatologists, 1 dual-trained neurologist-rheumatologist, and 1 patient representative. Neurologists were specifically chosen for SjD experience and collaboration with rheumatologists.
- Methodology drew on the ACR framework (plus ASCO, USPSTF, and ADA principles); recommendation strength was ultimately rated using GRADE.
- Systematic review: PubMed searched for English-language articles (Jan 1990 → May 2023, updated through 22 July 2025) and Embase through 22 July 2025. Screening, extraction, and appraisal were done in duplicate. The protocol was pre-registered (INPLASY 2025110020).
- Yield: 13,848 records → 11,174 after de-duplication → 10,918 excluded on title/abstract → 256 full-text assessed → 247 excluded → 9 studies included (6 case series, 1 retrospective cohort, 2 prospective cohorts, all without a comparison group). No study was eligible for meta-analysis.
- Consensus: An anonymous Delphi process (4 rounds; 74.3–95.7% participation) required ≥75% agreement. The Consensus Expert Panel (CEP) comprised 69 members — 39 rheumatologists, 20 neurologists, and 10 patients/family members.
- Conflict-of-interest policy: no more than 49% of TRG members could hold a direct COI; none had direct COIs and only 40% had indirect disclosures. Process adhered to AGREE II and PRISMA.
A key methodological honesty: because the underlying evidence was almost uniformly very low quality, most recommendations rest on expert consensus rather than trial data.
The Results
The guideline delivered three products: an aligned cross-specialty nomenclature, 31 good-practice statements for evaluation, and 20 treatment recommendations (14 conditional, 6 strong). Evidence quality was rated very low for almost all treatment recommendations. A striking feature is that strong recommendations were issued despite very-low-certainty evidence — permissible under GRADE when benefits clearly outweigh harms and inaction is unacceptable to patients.
The 6 Strong Recommendations (Worth Memorising)
- Neuropathic pain in any PNS setting → first-line gabapentin, pregabalin, or an SNRI (Rec 4).
- Acquired CIDP-type demyelinating polyradiculoneuropathy → treat as standard CIDP (Rec 9).
- Sensory ganglionopathy with significant symptoms → aggressive, immediate moderate-to-high-dose glucocorticoids plus immunomodulation — because the ataxia can be rapidly and irreversibly disabling (Rec 10).
- High suspicion of vasculitic neuropathy → high-dose glucocorticoids + rituximab as first line (rituximab preferred over cyclophosphamide for its safer profile) (Rec 11).
- Vasculitic neuropathy with persistent pain → symptomatic management per the neuropathic-pain recommendations (Rec 14).
- Autonomic neuropathy → patient education and non-pharmacologic/lifestyle measures (Rec 16).
Mononeuropathy Therapy (all very low / conditional)
- Trigeminal neuropathy → short oral glucocorticoid course if an acute inflammatory cause is suspected (Rec 1).
- Trigeminal neuralgia → carbamazepine/oxcarbazepine first-line; second-line/adjuncts include gabapentin, pregabalin, baclofen, lamotrigine, lacosamide, phenytoin (Rec 2).
- Acute facial neuropathy → glucocorticoids for ≥10 days, consider adding valacyclovir (Rec 3).
Polyneuropathy Therapy (beyond the strong ones above)
- Refractory neuropathic pain → sodium-channel blockers (carbamazepine, oxcarbazepine, lamotrigine) or TCAs (nortriptyline, desipramine better tolerated); refer if intolerant/refractory (Rec 5).
- Progressive/aggressive polyneuropathy → empiric glucocorticoid trial (Rec 6).
- Immune-mediated non-vasculitic large-fibre → consider IVIG (Rec 7).
- Immune-mediated small-fibre, severe/progressive and unresponsive to symptomatic therapy → IVIG ≥3 months (SCIG as alternative) (Rec 8).
- Vasculitic neuropathy unresponsive to GC+rituximab → add cyclophosphamide as second line (Rec 12).
- Vasculitic + cryoglobulinemia → maintenance immunomodulatory therapy (Rec 13).
- IVIG is NOT recommended first-line for vasculitic neuropathy (Rec 15).
Autonomic Therapy
- Orthostatic tachycardia → lifestyle first (fluids, salt, compression garments, graded seated/supine exercise; deprescribe hypotension/tachycardia-provoking drugs). For severe cases: non-selective beta-blockers or ivabradine (HCN-channel blocker) for rate control; midodrine/droxidopa or fludrocortisone/IV saline for low-BP-driven tachycardia; pyridostigmine may help stabilise HR/BP (Rec 17).
- Orthostatic hypotension → multidisciplinary assessment/management (Rec 18).
- Severe immune-mediated autonomic dysfunction → IVIG (Rec 19), then rituximab if IVIG fails (Rec 20).
Evaluation “Good Practices” — High-Yield Practical Points
- Always exclude alternative neuropathy causes: diabetes, hypothyroidism, B12 deficiency, monoclonal gammopathy, toxins, medications, infection.
- Small-fibre neuropathy → punch skin biopsy for epidermal nerve fibre density.
- NCS/EMG best timed 1–4 weeks from symptom onset; performed by an experienced electromyographer.
- Reserve lumbar puncture for suspected AIDP/CIDP, ganglionopathy, neoplastic disease, or infection.
- Red flags for vasculitic neuropathy: acute/painful/stepwise multiple mononeuropathies, plus cryoglobulinemia, hypocomplementemia, monoclonal gammopathy, positive RF, or raised inflammatory markers. Confirm with nerve + muscle biopsy; because serum cryoglobulins are technically difficult to detect, positive RF + low C4 can serve as surrogates.
- If carpal tunnel release is performed, consider tenosynovial biopsy to exclude amyloidosis.
- Autonomic work-up: COMPASS-31 questionnaire + postural vitals; formal testing (tilt-table, QSART, HRV, Valsalva); CASS score for severity; an autoimmune dysautonomia antibody panel (ACh-receptor ganglionic antibody, ANNA-1, CASPR2, CRMP-5, DPPX, LGI1, PCA-2, AP3B2). Autonomic failure + sensory ataxia should trigger urgent neurology referral for possible ganglionopathy.
Consensus strength: 18/20 recommendations and 30/31 good practices reached ≥90% agreement. The lowest agreement (79%) was for the strong label on IVIG for non-vasculitic large-fibre polyneuropathy (Rec 7) — driven by cost, side-effect, and evidence-quality concerns — which is presumably why it was ultimately framed as conditional.
Study Limitations
- No comparison-group studies — the entire evidence base is case series and uncontrolled cohorts, raising bias risk and limiting generalisability. No RCTs and no meta-analysis were possible.
- Very-low-certainty evidence underpins essentially all treatment recommendations; the document is candidly “predominantly expert opinion.”
- Evidence gaps for specific manifestations (hearing loss, vertigo, CN IX/X dysfunction) meant no recommendations could be made for these.
- Diagnostic-test performance data (sensitivity, specificity, predictive values, likelihood ratios) in an SjD-specific population were absent, forcing a “good practices” approach rather than evidence-graded diagnostic recommendations.
- Strong recommendations built on very-low evidence require careful, individualised, shared decision-making rather than reflexive application.
How This Adds to Practice
- It is broadly consistent with the 2020 EULAR ESSDAI-based recommendations (shared reliance on glucocorticoids, immunomodulators, biologics) but goes further by: aligning neuro-rheum nomenclature; giving localisation-specific guidance; explicitly addressing autonomic neuropathy, small-fibre disease, and neuropathic pain; and prioritising specific immunosuppressants.
- It foregrounds rituximab / B-cell-targeted therapy for severe or refractory disease (vasculitic neuropathy, treatment-resistant immune-mediated autonomic dysfunction) — timely given that two late-phase SjD trials recently met primary endpoints: ianalumab (anti-BAFF-receptor, phase 3 NEPTUNUS) and telitacicept (BLyS/APRIL dual inhibitor). Additional agents in trials include deucravacitinib, dazodalibep, and efgartigimod.
- It reframes the artificial “evidence-based vs consensus-based” divide (citing Djulbegovic & Guyatt): all guidelines require both evidence appraisal and expert consensus, and consensus is legitimate — even essential — where trial data are scarce.
- It offers a concrete screening symptom checklist (systemic, oral, ocular) to raise suspicion of underlying SjD in patients presenting with neuropathy.
- Planned review in 3–5 years; corneal neuropathy is being deferred to the forthcoming ocular guideline update.
Final Take-Aways
- Suspect PNS involvement early in SjD — neuropathy often precedes the diagnosis, may be the presenting feature, and can occur despite a negative anti-SSA.
- Characterise the neuropathy subtype (mono- vs poly- vs autonomic; large- vs small-fibre; vasculitic vs immune-mediated non-vasculitic) — because treatment diverges sharply by phenotype.
- Don’t miss vasculitic neuropathy or ganglionopathy — both are potentially rapidly disabling and warrant immediate aggressive immunotherapy (high-dose steroids + rituximab for vasculitis; steroids + immunomodulation for ganglionopathy).
- Manage neuropathic pain proactively (gabapentin/pregabalin/SNRI first-line) — one of the few “strong” recommendations, and standard of care.
- IVIG has a defined but limited role — reasonable for immune-mediated large- and small-fibre disease, but not first-line for vasculitic neuropathy.
- Autonomic dysfunction deserves structured work-up (COMPASS-31 → formal autonomic testing) and a lifestyle-first, escalate-if-needed approach.
- This is a low-certainty, consensus-driven document — apply it through shared decision-making, and interpret “consider” as a prompt to individualise.
- Care should be multidisciplinary — neurology, rheumatology, and primary care working together, with a shared vocabulary, is the central organising principle of the guideline.