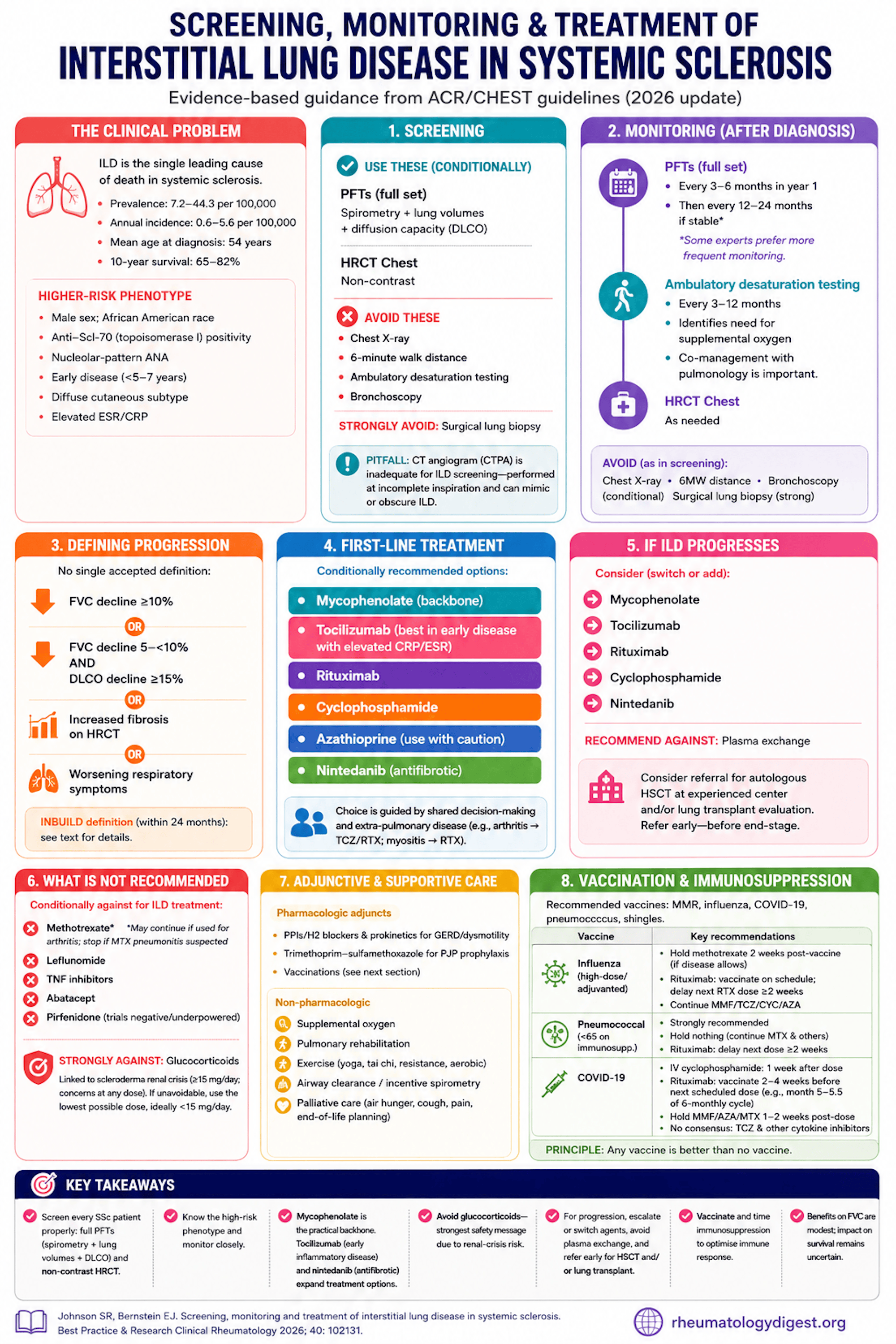
TL;DR: ILD is the leading cause of death in systemic sclerosis, so screen every patient properly (full PFTs + non-contrast HRCT — office spirometry and a CTPA don’t count); mycophenolate is the practical first-line backbone, with tocilizumab and nintedanib expanding the toolkit, while glucocorticoids carry the strongest recommendation against because of renal-crisis risk.
The Clinical Problem
Interstitial lung disease (ILD) is the single leading cause of death in systemic sclerosis (SSc), and a dominant driver of breathlessness, cough, fatigue, reduced exercise capacity, poor quality of life, and loss of employment.
The epidemiology frames the urgency:
- Prevalence 7.2–44.3 per 100,000; annual incidence 0.6–5.6 per 100,000.
- Mean age at SSc-ILD diagnosis 54 years.
- Ten-year survival 65–82%.
- Higher-risk phenotype: male sex, African American race, anti-Scl-70 (topoisomerase I) positivity, nucleolar-pattern ANA, early disease (within 5–7 years of first non-Raynaud symptom), the diffuse cutaneous subtype, and elevated acute-phase reactants (ESR, CRP).
The paper is essentially a practical distillation of the 2023 ACR/CHEST guidelines (screening/monitoring and treatment), supplemented by adjunctive measures and the ACR vaccination guidance. It is a guideline-translation review, not new primary data.
Screening — What to Use, What to Avoid
The right screen is PFTs + high-resolution CT (HRCT) chest, both conditionally recommended.
- PFTs must include spirometry, lung volumes, AND diffusion capacity (DLCO). Office spirometry alone is insufficient (no lung volumes or DLCO).
- HRCT must be non-contrast. A CT angiogram (CTPA) is inadequate for ILD screening — a non-obvious but important pitfall: CTPA is acquired at incomplete inspiration to maximise pulmonary-artery enhancement, generating atelectasis that can mimic or obscure ILD. So a “normal” PE study does not clear the lungs of ILD.
- Recommended against for screening: chest X-ray, 6-minute walk distance, ambulatory desaturation testing, bronchoscopy (all conditional). Strong recommendation against surgical lung biopsy.
- These “against” tests still have legitimate non-screening uses — e.g., bronchoscopy to exclude infection, sarcoidosis, lymphoma, or alveolar haemorrhage; surgical biopsy to exclude malignancy.
Monitoring Once ILD Is Diagnosed
- PFTs (full set): every 3–6 months in the first year, then every 12–24 months if stable. A practical caveat flagged by the authors: some experts consider 24 months too long and prefer more frequent PFTs even in apparently stable disease.
- Ambulatory desaturation testing every 3–12 months — conditionally recommended because it flags the need for supplemental oxygen (note: the 6-minute walk distance is not adequate for this; and desaturation testing usually sits outside rheumatology scope, so co-management with pulmonology matters).
- HRCT as needed.
- Same tests recommended against as in screening (X-ray, 6MWD, bronchoscopy conditional; surgical biopsy strong against).
Defining “Progression” — No Single Accepted Definition
Several are in use — worth knowing because trials and guidelines diverge:
- FVC fall ≥10% from baseline; OR
- FVC fall 5–<10% combined with DLCO fall ≥15%; OR
- increased radiographic extent of fibrosis on HRCT.
- The INBUILD definition (within 24 months): relative FVC decline ≥10% predicted; or FVC decline 5–<10% plus worsening symptoms or increased HRCT fibrosis; or worsening symptoms plus increased HRCT fibrosis.
First-Line Pharmacologic Treatment
Conditionally recommended options: mycophenolate, tocilizumab, rituximab, cyclophosphamide, azathioprine, nintedanib. Choice is driven by shared decision-making and extra-pulmonary disease — e.g., inflammatory arthritis favours tocilizumab/rituximab; myositis favours rituximab.
The trial evidence the authors marshal:
- Mycophenolate (the backbone): In Scleroderma Lung Study II (SLS II), MMF and cyclophosphamide gave near-identical modest FVC improvement (MMF +2.19%, CYC +2.88%; p=0.24) — but MMF had less leukopenia/thrombocytopenia and is cheaper with more clinical experience, which is why it is preferred. The Naidu pilot vs placebo showed a similar small effect size.
- Cyclophosphamide: SLS I showed a modest but significant adjusted 12-month FVC benefit vs placebo (+2.53%, p<0.03), at the cost of more leukopenia/neutropenia. The Hoyles (IV CYC → azathioprine) trial gave a comparable non-significant FVC trend (+4.19%, p=0.08).
- Azathioprine — use with caution: the Nadashkevich open-label trial showed FVC worsening on azathioprine (91.7% → 83.4%, p<0.01) while cyclophosphamide held steady. Hence experts urge caution with AZA for ILD specifically.
- Rituximab: The Indian RCT (Sircar) showed RTX improved FVC (61.3% → 67.5%) while CYC declined (p=0.003). DESIRES (Japan) was skin-score-primary but showed an FVC advantage (+2.96% difference, p=0.044) and supported RTX safety. RECITAL (RTX vs CYC) found both improved FVC with no superiority of RTX over CYC (difference −40 mL, p=0.49).
- Tocilizumab: Both faSScinate (phase 2) and focuSSced (phase 3) missed their primary skin-score endpoints but showed preservation of FVC — focuSSced FVC difference +4.2% (p=0.0002). This FDA-approved tocilizumab for SSc-ILD, with the strongest signal in early disease with elevated acute-phase reactants. Watch for serious infections.
- Nintedanib (antifibrotic): SENSCIS — the largest SSc-ILD trial (n=576) — slowed FVC decline (−52.4 vs −93.3 mL/year; difference 40.9 mL/year, p=0.03). Adding nintedanib to MMF gave numerically greater preservation but not significant (and the trial was not powered for it). Tolerability matters: diarrhoea 75.7%, weight loss, and ALT/AST ≥3× ULN in ~4.9%.
Progression Despite First-Line Therapy
Switch or add among mycophenolate, tocilizumab, rituximab, cyclophosphamide, nintedanib. Recommend against plasma exchange. Consider referral for autologous HSCT at an experienced centre and/or lung transplant evaluation — and time the transplant referral before disease is end-stage.
What Is NOT Recommended
- Conditionally against (for ILD treatment): methotrexate, leflunomide, TNF inhibitors, abatacept. Caveat: a recommendation against MTX for ILD does not mandate stopping MTX used for arthritis — but stop it if MTX pneumonitis is suspected.
- Pirfenidone: the Acharya pilot and SLS III (MMF + pirfenidone) were both negative/underpowered (SLS III FVC difference p=0.93, with more GI adverse events). Not recommended.
- Glucocorticoids — STRONG recommendation against, the standout safety message. Steroids are linked to scleroderma renal crisis, classically at ≥15 mg/day, with concern at any dose. If unavoidable, use the lowest possible dose, ideally <15 mg/day.
- Nerandomilast (PDE4B inhibitor, FIBRONEER-ILD) reduced FVC decline vs placebo but was published after the guideline and is not yet incorporated — one to watch.
Adjunctive (Non-Drug and Supportive) Care — Often Forgotten
- Pharmacologic adjuncts: PPIs/H2 blockers and prokinetics (domperidone) for reflux/dysmotility; trimethoprim-sulfamethoxazole for PJP prophylaxis; vaccinations.
- Non-pharmacologic: supplemental oxygen, pulmonary rehabilitation, graded exercise (yoga, tai chi, resistance, aerobic), airway clearance/incentive spirometry, and palliative care for air hunger, cough, pain, and end-of-life planning.
Vaccination + Immunosuppression Timing
Recommended vaccines: MMR, influenza, COVID-19, pneumococcus, shingles. The guiding principle: any vaccine beats no vaccine, but immunogenicity can be optimised by timing drug holds:
- Influenza: prefer high-dose/adjuvanted. Hold methotrexate 2 weeks post-vaccine (if disease allows). Rituximab: vaccinate on schedule and delay the next RTX dose ≥2 weeks. Continue MMF/TCZ/CYC/AZA.
- Pneumococcus: strongly recommended in those <65 on immunosuppression. Hold nothing (continue MTX and others); for RTX, delay next dose ≥2 weeks.
- COVID-19: IV cyclophosphamide 1 week after each dose; rituximab — vaccinate 2–4 weeks before the next scheduled dose (e.g., month 5–5.5 of a 6-monthly cycle); hold MMF/AZA/MTX 1–2 weeks after each dose. No consensus on interrupting tocilizumab/other cytokine inhibitors.
Key Takeaways
- Screen every SSc patient properly: full PFTs (spirometry + lung volumes + DLCO) and non-contrast HRCT. Office spirometry alone and a CTPA do not count.
- Know the high-risk phenotype (diffuse cutaneous, anti-Scl-70, male, early disease, raised CRP/ESR) — these patients warrant the closest surveillance.
- Monitor on a schedule: full PFTs every 3–6 months in year 1, then 12–24 months if stable (many experts monitor more often); add ambulatory desaturation testing and HRCT as needed — and co-manage with pulmonology.
- Mycophenolate is the practical backbone (efficacy comparable to cyclophosphamide, better tolerated, cheaper); tocilizumab (FDA-approved, best in early inflammatory disease) and nintedanib (antifibrotic, slows FVC decline) meaningfully expand the toolkit. Be cautious with azathioprine.
- Let extra-pulmonary disease guide drug choice through shared decision-making.
- Avoid glucocorticoids — the strongest safety statement, because of renal-crisis risk; if used, keep <15 mg/day.
- For progressive disease, escalate/switch agents, avoid plasma exchange, and refer early for HSCT and/or lung transplant evaluation.
- Vaccinate and choreograph immunosuppression around vaccination to maximise immunogenicity.
- Honest limitation: the FVC benefits across these agents are modest, and their effect on survival remains uncertain — the authors call for head-to-head combination trials and biomarkers to guide therapy.